

A simulation of a solvated nanoparticle, using our algorithm for carrying out molecular dynamics simulations without the use of periodic replicas of the system.
Research in the Gezelter lab centers on understanding how complex physical behavior emerges from simple interactions between molecules. The primary tool we have for exploring emergent behavior is molecular simulations. Most of our work involves creating new algorithms that allow computers to probe the dynamics of chemical systems in interesting ways. We apply our new methods to study dynamics and transport at the boundaries between dissimilar materials and phases of matter. Simulations are complemented by analytical statistical mechanics and by collaboration with other experimental and theoretical groups.
Much of our work deals with creating algorithms that allow computers to probe chemical systems in ways that were not previously possible. Of particular interest are:
- shear-flow separation of enantiomers,
- electrostatic interactions in condensed phases,
- reverse non-equilibrium molecular dynamics (RNEMD) methods that drive simulations into unstable, but steady-state regimes, enabling the measurement of mass and energy transport on a nanometer scale, and
- methods to simulate chemical environments in implicit solvent environments.
Although our new methodology can be applied quite generally, it was created to solve particular problems. We are generally involved in dynamical studies of how interfaces change in response to perturbations. These systems include:
- Ice / water interfaces
- Surface reconstruction of catalytic metal surfaces
- Metal nanoparticle interfaces
- Liquid crystals and field-induced phase transitions
Shear Flow Separation of Enantiomers
One of our newest projects involves the discovery of a geometric property of molecules that has a direct analogy to the pitch of a screw. Enantiomers of chiral molecules are nonsuperposable mirror images with the same structural formula. They are generally difficult to separate without significant expense and lost material. The molecular pitch is a property that can be used to separate these molecules in a rotating or shearing fluid. Learn more about this research at our shear-flow separation page.
Real-space Electrostatics
In a computer simulation of biological molecules in aqueous environments, the processor spends roughly 90% of its time computing the electrostatic interactions between atoms or molecular substructures, and relatively little time on local or bonded interactions. Electrostatics are the dominant force in chemistry, and a large component of my research program is concerned with making computation of this interaction faster and easier to understand. We have become involved in recent advances in real space electrostatic methods, which make a set of simple assumptions about how charges, dipoles, and higher order multipoles are organized in chemical systems. Our work on damped shifted force (DSF) electrostatics has become widely used, and is now available in popular molecular simulation packages like OpenMD, DL_POLY and LAMMPS.
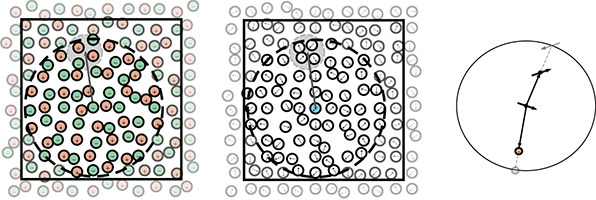
Ionic materials exhibit local clustering of dissimilar charges (in the smaller grey circle), so interactions are weakened at large distances. Motion of individual ions in and out of a cutoff sphere (larger dark circle) can break the multipolar clustering. Dipolar fluids (middle) exhibit similar clustering. Placement of reversed image charges or image multipoles on the surface of the cutoff sphere (right) recovers the effective short-ranged behavior.
Our most recent papers are a collaboration that involves physicist Kathie Newman (also at Notre Dame). This work includes extensions to the damped shifted force technique for molecular dipoles and quadrupoles. Because the real-space methods are fast and accurate, they are a promising way to handle complex fluids in computer simulations.
Non-equilibrium molecular dynamics
In bulk materials, transport properties like thermal conductivity, viscosity, and diffusion are straightforward to compute using standard simulation techniques. When the energy or mass transport occurs at the interface between dissimilar materials, the traditional approaches become much more difficult.
It is possible to force a computer model into a non-equilibrium state and to let the response to the non-equilibrium conditions provide evidence for the transport properties. In early approaches to non-equilibrium molecular dynamics (NEMD), a physical (e.g., a temperature, velocity or concentration) gradient was imposed on the computer model, and the transport properties (conductivity, viscosity or diffusivity) are deduced by watching the flow in the system as it tries to return to equilibrium. However, at interfaces between dissimilar materials, it was not always clear what kind of gradient should be applied. Reverse Non-Equilibrium Molecular Dynamics (RNEMD) methods flip this technique around and impose an unphysical energy flux between different regions of the computer model. The model then responds by developing a temperature, momentum, or concentration gradient between the two regions. The transport properties (including interfacial transport coefficients) are then easily deduced using linear response theory.
We have developed three novel RNEMD methods specifically tailored to understand transport phenomena at heterogeneous interfaces. Using these new algorithms, it is now possible for simulations to measure thermal conductivity (and interfacial thermal conductance), shear viscosity (and interfacial friction), bulk diffusion (and interfacial permeability). The most recent generalization of this work has included non-periodic simulation cells so that the same physical properties can be predicted for the nanometer-scale curved interfaces that are exhibited by nanoparticles.
Implicit Environments using Langevin Dynamics
We have a significant research footprint in Langevin dynamics, a simulation methodology which treats part of the solvent environment implicitly in the equations of motion rather than simulating the solvent atoms directly. On the molecular scale, we developed an algorithm for carrying out simulations on complex molecular bodies by incorporating the hydrodynamic resistance tensors into an advanced rotational integration scheme. This allows us to simulate large structures like proteins and nanoparticles that are moving through a solvent without actually including atoms from the solvent.
For large molecular structures (e.g. chemically-protected solvated nanoparticles) we developed a new isobaric-isothermal (NPT) algorithm which applies an external pressure to a convex hull surrounding the system. A Langevin thermostat is also applied to the facets to mimic contact with an external heat bath. This new method, the Langevin Hull, can easily handle heterogeneous mixtures of materials with different compressibilities, and has proven extremely useful to study heat transport at the surfaces of nanoparticles of various shapes (e.g. spheres, icosahedra, and pentagonal multiply-twinned rods).
Ice / Water interfaces
We have long-standing interests in the different crystalline morphologies of ice and ice/water interfaces. One of the early discoveries in the group was a new crystal structure for ice that was predicted via computer simulations, but which has not yet been seen in experiments, We have lately been using our RNEMD methodology to study the hydrophobicity/hydrophilicity of the different facets of regular ice Ih. Learn more about this research at our ice/water webpage.
Surface reconstruction and classical approaches to multiple oxidation states
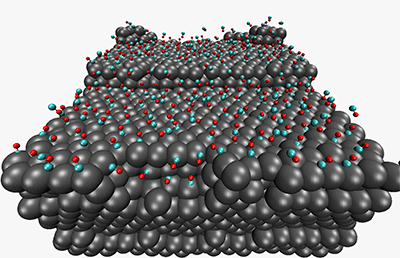
A reconstructed Pt(557) surface after 86 ns exposure to a half a monolayer of CO.
We have looked previously at the morphological changes that take place at catalytic surfaces (e.g. Pt(557)) in the presence of reactant gas (carbon monoxide) overpressure. Until recently, molecular dynamics has only been able to observe non-reactive physical changes at interfaces. One of the directions we are taking our electrostatics work is into fluctuating-charge (fluc-q) models for polarizability and oxidation. Of particular interest is modeling the multiple oxidation states exhibited by metal atoms at metal-oxide interfaces. We are developing multiple-minimum fluctuating charge models which predict the dynamics of oxide layer growth at metal surfaces.
Metal nanoparticle interfaces
At the surfaces of metal nanoparticles, the chemical bond between the metal and the ligand introduces vibrational overlap that is not present between the bare metal surface and solvent. We have been able to show that the presence of a partial-monolayer ligand coverage yields a higher interfacial thermal conductance value than the bare nanoparticle. Learn more about this research at our nanoparticle heat transport webpage.
Liquid crystals and field-induced phase transitions
We have continuing interest in molecules which exhibit multiple liquid-like phases with different internal arrangements of the molecules. 4-cyano-4′-pentylbiphenyl (5CB) is a liquid crystal forming compound with a terminal nitrile group aligned with the long axis of the molecule. We observed that an isotropic-nematic phase transition can be induced by the application of an electric field, and that the terminal nitrile group exhibits a ~2.5 cm-1 red shift as a result of the phase transition. Our simulations indicate that the phase transition opens up space around the nitrile bond, and that this “anti-caging” contributes to the change in the nitrile spectrum.
We also have worked on coarse-grained models of lipids and have made predictions about the structure of the elusive Pβ‘ rippled phase of lipid bilayers. This phase has excited substantial experimental interest over the past 4 decades, and our molecular-scale models suggest a role for the head-group packing and orientational ordering in creating the nanometer-scale buckling of these important membranes.